
Una nuova variante emoglobinica, denominata “Emoglobina Monza”, è stata individuata da un gruppo di ricercatori dell’Università di Milano-Bicocca.
Questa variante, causata da una duplicazione di 23 aminoacidi nel gene dell’emoglobina (HBB), comporta instabilità della proteina, provocando episodi di anemia emolitica acuta, soprattutto in occasione di episodi febbrili. La scoperta, pubblicata sulla rivista Med di Cell Press, apre il campo a nuove prospettive per lo studio di queste rare patologie grazie all’utilizzo di tecniche di Intelligenza artificiale.
Il caso clinico che ha portato alla scoperta è stato quello di una bambina di origine cinese che, a seguito di un episodio febbrile, ha sviluppato una grave anemia emolitica, condizione patologica caratterizzata da una distruzione accelerata dei globuli rossi, a un ritmo superiore alla capacità del midollo osseo di produrli. Le conseguenze della patologia possono essere gravi, specialmente in età pediatrica quando gli episodi acuti possono compromettere seriamente lo stato di salute dei piccoli pazienti. «Esistono varianti emoglobiniche, note come “emoglobine instabili”, che tendono a essere degradate (ovvero distrutte) sotto stress fisici, come gli episodi febbrili, scatenando così crisi emolitiche. A causarle generalmente sono alterazioni puntiformi nella sequenza amminoacidica dell’emoglobina, che modifica la stabilità e la funzionalità della proteina stessa», spiega Carlo Gambacorti-Passerini, direttore del reparto di Ematologia della Fondazione IRCCS San Gerardo dei Tintori di Monza e professore presso l’Università di Milano-Bicocca, che ha coordinato il progetto di ricerca.
La piccola paziente è stata seguita presso la Fondazione IRCCS San Gerardo dei Tintori di Monza dalla pediatra Paola Corti e dal tecnico Amedeo Messina, che hanno realizzato che l’anemia era dovuta a una variante anomala di emoglobina con un comportamento instabile in situazioni di stress. Indagini successive hanno rivelato che anche la madre e i due fratelli della bambina possedevano la stessa variante e manifestavano episodi simili nel corso di episodi febbrili. Un’analisi genetica specifica ha mostrato che la variante non solo era inedita, ma era anche caratterizzata da una duplicazione molto lunga (23 aminoacidi) del gene che codifica la catena beta dell’Emoglobina (HBB), una caratteristica mai osservata prima in altre emoglobine instabili.
Le duplicazioni lunghe nel gene HBB sono molto rare e sono state sempre associate a un’altra malattia, la beta-talassemia. Infatti, si è sempre ritenuto che le lunghe duplicazioni comportino un’alterata interazione tra le due catene che compongono l’emoglobina, Beta e Alfa. Il dottor Ivan Civettini, ematologo, ora dottorando presso l’IRCCS Ospedale San Raffaele, e la dottoressa Arianna Zappaterra, medico presso la divisione di Ematologia della Fondazione IRCCS San Gerardo dei Tintori di Monza, si sono quindi chiesti come una mutazione di tale portata potesse comunque consentire all’emoglobina di mantenere una funzionalità normale, almeno in condizioni fisiologiche.
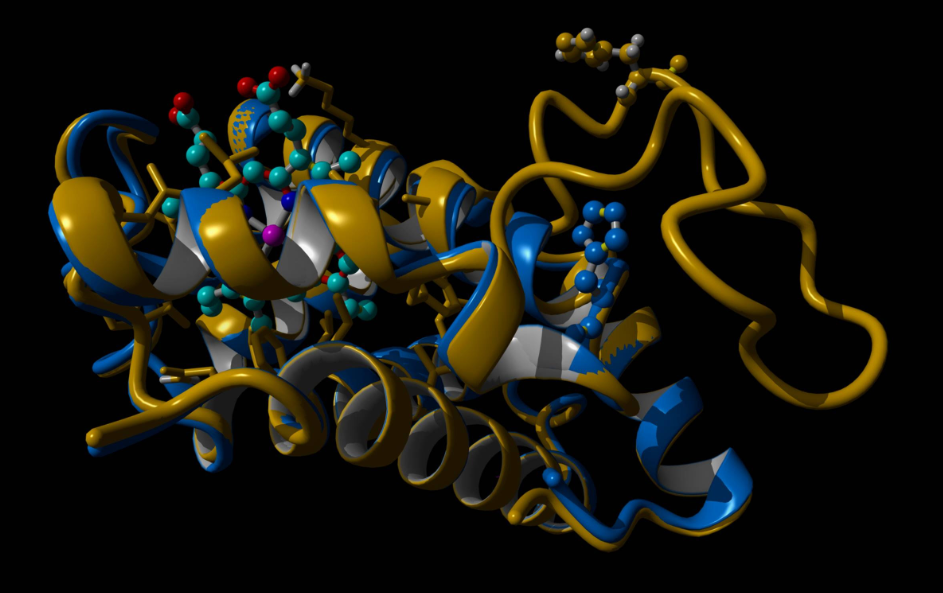
«La struttura della variante emoglobinica è stata ricreata utilizzando tecniche di modeling tridimensionale e intelligenza artificiale (reti neurali), recentemente premiate con il Nobel per la chimica», precisa Ivan Civettini. «In condizioni normali, il legame tra le due catene dell’emoglobina è preservato e la duplicazione si presenta come una lunga protrusione che sbatte un po’ come una banderuola nel vento, al di fuori della struttura proteica dell’emoglobina. Inoltre abbiamo osservato che questa mutazione non compromette il centro attivo dell’emoglobina, dove avviene il legame con ossigeno e ferro. In sintesi, in condizioni normali, l’emoglobina Monza resta stabile e il legame preservato tra le catene dell’emoglobina non causa beta-talassemia».
Che cosa avviene dunque nel sangue durante l’episodio febbrile? Per ricreare questa condizione sono state utilizzate ulteriori tecniche computazionali avanzate, note come “dinamica molecolare”. È stato ricreato un fluido con la stessa “salinità” del sangue umano, dove è stata inserita l’emoglobina normale e l’emoglobina Monza e che è stato portato alla temperatura di 38°C, come durante un episodio febbrile. Risultato? L’Emoglobina Monza si degrada più velocemente di quella normale, perdendo il contatto con l’atomo di ferro. Questi esperimenti sono stati eseguiti in collaborazione col professor Alfonso Zambon dell’Università di Modena e Reggio Emilia.
«La scoperta offre nuovi spunti per comprendere meglio varianti rare di emoglobina, ma che diverranno sempre più frequenti in Italia con l’aumento di etnie diverse da quella caucasica», aggiunge Carlo Gambacorti-Passerini. «L’uso di tecniche computazionali moderne e l’ausilio dell’intelligenza artificiale hanno reso questo tipo di studi più rapido ed economico rispetto a metodi tradizionali come, per esempio, la cristallografia a raggi X. Un’ulteriore prova dell’importanza della collaborazione tra diverse istituzioni nella medicina moderna».